Technologies
RNA sequencing
Typically, we isolate total RNA employing the RNeasy Mini kit (Qiagen) including Trizol treatment, but the exact choice of the kit depends on the tissue to analyse. The quality and amount of the RNA are assessed by NanoPhotometer (Implen), Qubit Fluorometer (Invitrogen) and by an Agilent 2100 Bioanalyzer.
Sequencing is done in our in-house core facility or by external partners typically using a paired-end stranded protocol resulting in 5-10 Gb of data per sample. Standard pipelines require an input of >100 ng pure intact total RNA. In case the amount of RNA is limited, other kits starting from at least 100 pg of total RNA may be used.
Data Analysis
Our RNAseq pipeline provides tools for intensive quality control of the raw data. Reads are then aligned to the genome and gene-level read counts are generated. We exclude low expressed genes from the dataset and determine significantly regulated genes with deseq2. For data visualization we provide Principal Component Analyses (PCAs), heatmaps and other representations. Detailed analysis settings and the list of utilised R packages are provided for each project. Normalised count data, gene annotation and statistics are summarized in an Excel file. To address the biological interpretation of the observed gene regulation we perform pathway and other enrichment analyses through the use of QIAGEN’s Ingenuity Pathway Analysis software (IPA®, Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014 Feb 15;30(4):523-30) and by g:profiler (https://biit.cs.ut.ee/gprofiler/gost). These analyses can be used directly for publications or might aid in the generation of new hypothesis, which can be followed up at the bench. Examples from an IPA analysis are shown below, with colors indicating observed gene regulation and predicted gene activation.
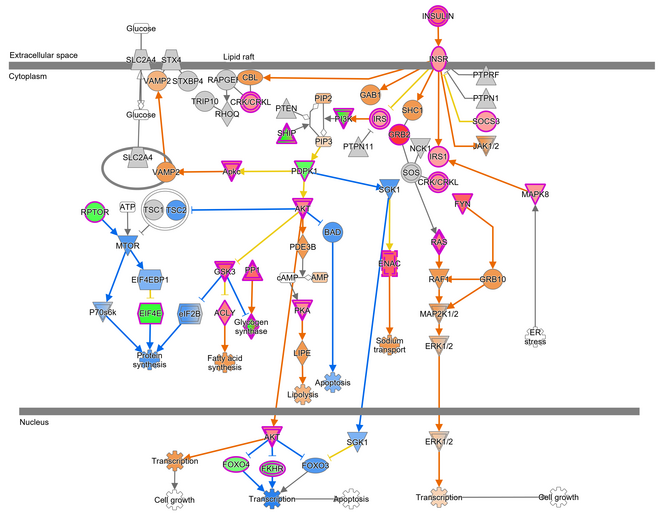
Source: GMC
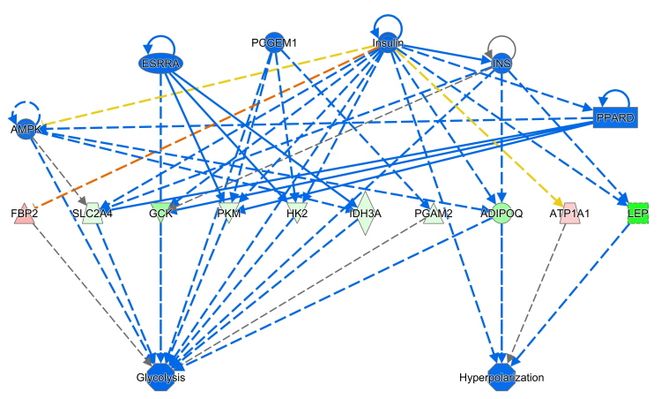
Source: GMC